An introduction to rstoolbox¶
Heuristic approaches to protein computational design have become extremely prolific due to potentially unlimited size of the sequence-structure space. Thus, computational design platforms such as Rosetta does not tend to generate a single optimal design but a population of decoys amongst which biologically relevant designs are expected to appear. As a matter of fact, depending on the size of the search space, the number of necessary decoys can range from one up to a million, as it is summarised in this image from the Rosetta documentation page:
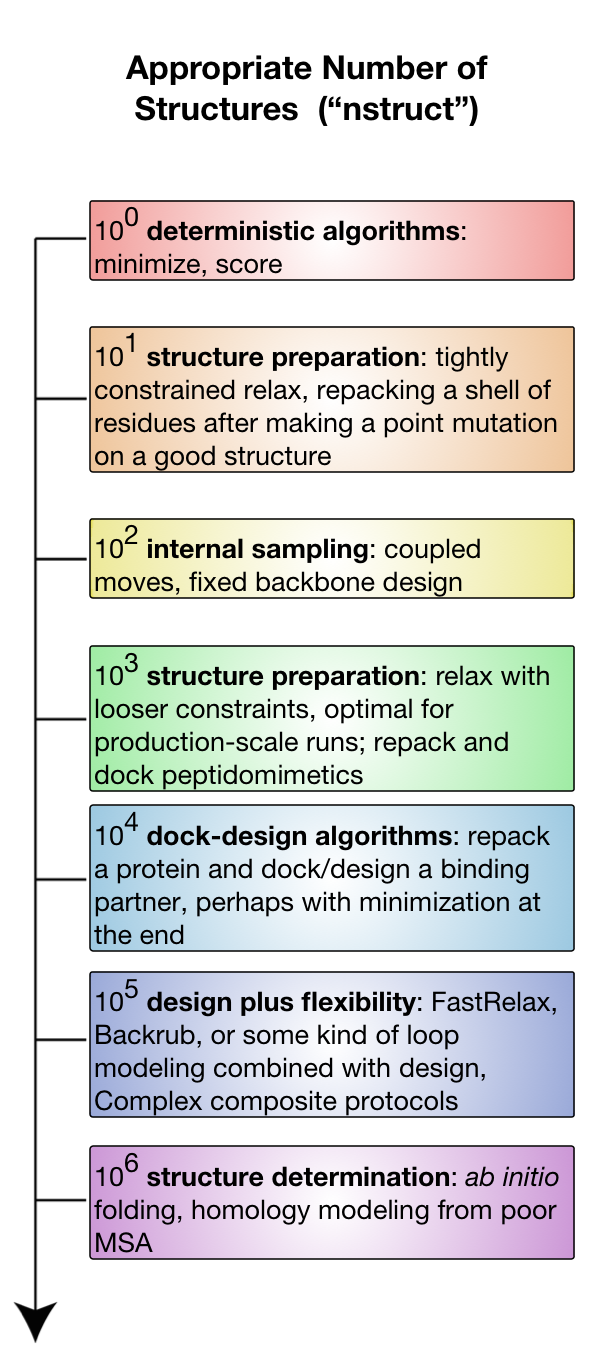
The rstoolbox aims to analyse these decoy populations in order to aid in the selection of a manageable number of decoys for experimental validation.
Furthermore, is can help protocol developers to benchmark their data in order to guide the development of new design approaches.
Although mostly prepared to work with Rosetta, most of the tools integrated in the rstoolbox would work with any other design system generating big decoy populations
by simply adding the appropriate parser.